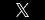


 by HWC Partner Carter Clay
by HWC Partner Carter Clay
To establish the existence of a safer design, a plaintiff, typically through an expert must provide substantial evidence to show (1) that the injuries caused by the product would have been less severe or eliminated by the use of an alternative design and (2) that the utility of the alternative design outweighed the utility of the design actually used. General Motors Corp. v. Jernigan, 883 So. 2d 646, 662 (Ala. 2003). There is no rule of Alabama law that states that the expert must test the safer alternative design to meet the substantial evidence threshold. In fact, “[no] one denies that an expert might draw a conclusion from a set of observations based on extensive and specialized experience.” Kumho Tire Co., Ltd. V. Carmichael, 526 U.S. 137, 156 (1999).
I. Examples of Substantial Evidence